血小板無力症(けっしょうばんむりょくしょう、thrombasthenia)あるいはグランツマンの血小板無力症(Glanzmann's thrombasthenia; GT)は、血小板の機能異常によって、粘膜や皮膚の出血が止まりにくくなり、出血傾向を来たす疾患である。先天性血液凝固障害のひとつで、常染色体劣性遺伝の遺伝形式をとる。
概要
1918年、グランツマン(Glanzmann)によって、血小板数が正常にもかかわらず出血傾向を生じる疾患として報告された。報告時の疾患名はhereditary hemorrhagic thrombastheniaで、直訳すると遺伝性出血性血小板無力症である。
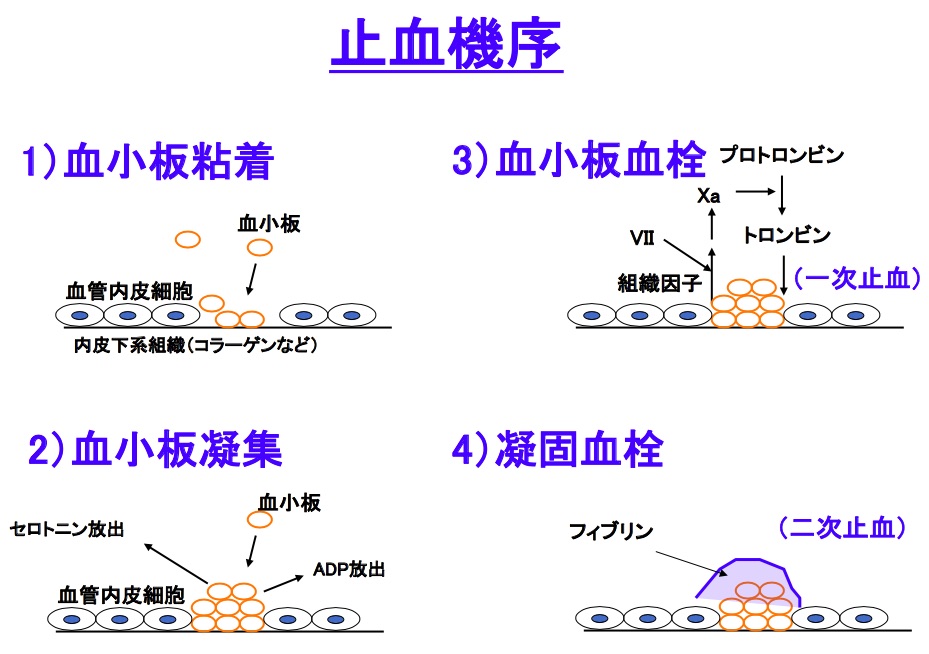
血小板無力症は、出血時の止血に重要な役割を果たしている血小板の機能異常によっておこる。血小板は、血管の傷害が起こると、露出した血管内皮細胞のコラーゲンに反応して粘着・凝集する。この凝集に関わる分子、GPⅡb/Ⅲa(αⅡbβ3インテグリン)が血小板上において欠損していることによって、血小板が凝集できなくなる。血小板機能(質)の異常であるため、血小板数(量)は一般に正常である。
GPⅡb/Ⅲa(αⅡbβ3インテグリン)の欠損は遺伝子異常によるもので、常染色体劣性遺伝の遺伝形式をとる。この遺伝子異常は、GPⅡb/Ⅲaの量的異常と機能異常(質的な異常)に分けられる。症状としては、幼少時より鼻や歯肉からの出血、女性においては月経出血の増加など、皮膚粘膜出血が主で、ほかの血液凝固障害でよくみられる関節内出血は一般には認められない。日本においては、児童福祉法に定める小児慢性特定疾病に指定されている。
病態
正常な血小板は、ほかの血小板や細胞と結合するのための受容体として、インテグリンと呼ばれる糖タンパク質(GP)をもっている。血小板は血管の傷害が起こると、露出した血管内皮細胞のコラーゲンに、直接あるいはヴォン・ヴィレブランド因子を介し反応して粘着する。粘着によって血小板が活性化すると、GPⅡb/Ⅲa(αⅡbβ3インテグリン)と呼ばれる糖タンパクが活性化し、ここにフィブリノーゲンが結合する。フィブリノーゲンは血小板同士を接着する糊のような役割を果たしており、ここにさらに血小板が結合することによって凝集し、血栓を形成する。血小板が正常に機能すれば、この血栓によって出血は止めることができる。血液が凝固したとき、血小板とフィブリンは赤血球などの他の血球成分も取り込んで固まり血餅となる。この血餅が形成されたのちは、血小板内の収縮タンパク質によって収縮される血餅収縮と呼ばれる現象を起こす。
血小板減少症では、遺伝子の異常によって、血小板表面のGPⅡb/Ⅲa(αⅡbβ3インテグリン)が量的に減少あるいは機能異常を起こしている。GPⅡb/Ⅲaの欠損した血小板では、本来は血小板粘着後におこる扁平・伸展化といった形態変化は低下し、さらに血小板凝集が起こらなくなる。typeⅠやvariant型に分類される血小板減少症では、血餅退縮の欠如がみられることがある。
原因遺伝子
GPⅡb/Ⅲa(αⅡbβ3インテグリン)は、αⅡbと、β3の2つのサブユニットから構成されている。正常状態では血小板1個あたり約4万個が存在し、血小板上では膜糖タンパク質の約20%を占めるもっとも多いインテグリンである。これらを生成する遺伝子はαⅡb遺伝子とβ3遺伝子と呼ばれ、ともに17番染色体の長腕(q)に存在し、血小板のもととなる巨核球において発現する。血小板無力症では、これらの遺伝子産物であるαⅡbと、β3のサブユニットは両方とも顕著に減少している。これは、GPⅡb/Ⅲa(αⅡbβ3インテグリン)が巨核球において生成されるときに、まずαⅡbのもととなる前駆体proαⅡbとβ3が複合体を形成し、その後にゴルジ体に移行してproαⅡbが切断を受け、さらに複合体が糖鎖修飾を受けることによって完成し、ここで初めて膜表面へと発現するからである。つまり、αⅡb遺伝子とβ3遺伝子のどちらか一方のみが異常である場合でも、両方がそろわないと膜表面には発現しないため、両方が欠損することになる。
分類
おもにGPⅡb/Ⅲa(αⅡbβ3インテグリン)の量的異常と、機能異常に分類することができる。
- 量的異常
- typeⅠ - GPⅡb/Ⅲaの発現量が5%以下
- typeⅡ - GPⅡb/Ⅲaの発現量が20%以下(およそ10~20%)
- 機能異常
- variant型 - GPⅡb/Ⅲa量は正常と変わらない(50%以上)が機能に異常
それぞれの頻度は、typeⅠが78%、typeⅡが14%、variant型が8%とされている。
量的異常
遺伝子変異の種類として、typeⅠはナンセンス突然変異やフレームシフト突然変異などが生じることによって伝令RNA(mRNA)が喪失したり、mRNAスプライシングの異常によって生じる。typeⅡはミスセンス突然変異によって生じる。前述のように、変異する遺伝子はαⅡb遺伝子あるいはβ3遺伝子であり、片方のみの異常でも血小板膜上に複合体は発現しない。typeⅠとtypeⅡの変異の結果としては、αⅡbまたはβ3のいずれかのmRNA生成の異常、αⅡbまたはβ3の複合体形成部位の異常による複合体形成不全、αⅡb,β3またはproαⅡbとβ3複合体の細胞内輸送障害の3種類が考えられる。
typeⅠでは血餅退縮が欠如し、血小板内フィブリノーゲンが減少しているが、typeⅡでは血餅退縮や血小板フィブリノーゲン量はほぼ正常に近い。ただし、現存するGPⅡb/Ⅲa量と臨床症状との相関関係は明らかになっていない。
機能異常
血小板減少症は常染色体劣性遺伝の遺伝形式をとるため、両親が患者ではなく異常遺伝子の保因者である場合、少なくとも正常に比べ50%に減少している(右図参照)。保因者には出血傾向は認められないため、正常な血小板機能の発現には50%のGPⅡb/Ⅲa量で足りると考えられている。したがって、50%のGPⅡb/Ⅲa量があるにもかかわらず血小板無力症の症状が出る場合、GPⅡb/Ⅲaの機能異常(質の異常)であると考えられる。このタイプをvariant型と呼ぶ。
variant型では、GPⅡb/Ⅲa(αⅡbβ3インテグリン)は血小板膜上に発現している。機能異常の種類によって血小板内フィブリノーゲン量、血餅退縮、出血傾向の程度はまちまちである。機能異常には、結合部位の障害、αⅡbβ3の活性化の障害などが報告されている。
疫学
日本では、1986年の調査で222例が報告されている。その後、血小板無力症に対する理解が深まると、診断が容易になり多くの施設で経験されるようになったため、正確な頻度は得られにくくなっているとされる。ただし、稀な疾患であることには変わりないと考えられている。日本においては、児童福祉法に定める小児慢性特定疾病に指定されており、一定の条件を満たすと治療において公費による助成が行われる。
遺伝形式が常染色体劣性遺伝であるため、血族結婚が認められる家系も多い。民族内の血族結婚が多発するイスラエルにおける患者遺伝子解析では、イラク系ユダヤ人の系統ではβ3遺伝子の変異が、アラブ系ユダヤ人の系統ではαⅡb遺伝子の変異が存在することが示唆されている。
症状
症状の多くは生後から出現する。鼻粘膜や口腔粘膜などの皮膚表層の出血が主体となっており、鼻や歯肉からの出血、紫斑が生じ、ときに消化管からの出血や血尿が生じる。打撲などの外傷により頭蓋内出血など重篤な出血を生じることもある。初潮開始以降の女性では、月経過多などの頻度も高くなる。また、抜歯など医療処置の際、止血困難で診断されることもある。
一方、ほかの血液凝固障害でよく認められる関節内出血や筋肉内出血などの深部出血はほとんどみられない。
検査・診断
血小板数、血小板形態には異常がない。血小板による一次止血を調べる出血時間検査では、出血時間の延長がみられる。血餅収縮検査では、typeⅠとvariant型の一部では血餅収縮能の低下がみられ、typeⅡとvariant型の一部では正常となる。血小板機能検査では、アデノシン二リン酸(ADP)、コラーゲン、アドレナリン(旧称エピネフリン)、トロンビンなどの血小板凝集を引き起こす物質に対する凝集が欠如する。ただし、抗生物質で血小板凝集作用をもつリストセチンでは凝集する。そのほか特殊な検査法として、フローサイトメトリーによる検査法や、遺伝子の変異を直接調べる遺伝子検査などがある。病理検査においては、指穿刺で採取した末梢血塗抹標本において,凝集せず単在する血小板が特徴的である。
診断においては、凝固系および凝固因子の検査には異常を認めないことなどが特徴的となる。区別しなければいけない似た疾患として、ヴォン・ヴィレブランド病や、無フィブリノーゲン血症などがある。また、後天性のものとして似た症状が起こるものには、抗血小板療法やGPⅡb/Ⅲaに対する自己抗体の産生などがある。
治療・予後
出血や出血予防のための対症療法が基本で、血小板製剤による血小板輸血がもっとも効果的である。特に止血困難になったときや、外科的処置の際には血小板輸血を行う。また、鼻出血や口腔内出血などの粘膜出血には、血栓の分解を抑える抗プラスミン剤が有効である。根治療法としては、骨髄移植によって治癒することが可能である。しかし治療以前に、患者本人が出血の危険性を理解し、出血に結びつくことを避ける日常生活を過ごすことが大切である。
重要臓器からの出血がない限り、予後は良好である。日本における調査では、出血による死亡率は1976年には6.8%であったが、1991年には4.9%となっており、これは血小板輸血による止血管理が向上したことが原因と考えられている。
出典
参考文献
- 池田康夫, 丸山征郎「冨山佳昭「血小板無力症」」『血小板生物学』メディカルレビュー社、2004年。ISBN 4-89600-724-7。国立国会図書館書誌ID:000007369992。https://ndlsearch.ndl.go.jp/books/R100000002-I000007369992。
- 池田康夫, 丸山征郎「八木秀男・藤村吉博「von Willebrand因子」」メディカルレビュー社、2004年。ISBN 4-89600-724-7。国立国会図書館書誌ID:000007369992。https://ndlsearch.ndl.go.jp/books/R100000002-I000007369992。
- 浅野茂隆, 池田康夫, 内山卓, 大野仁嗣, 三輪史朗『三輪血液病学』(第3版)文光堂、2006年。CRID 1130282269710552704。ISBN 4830614196。国立国会図書館書誌ID:000008056173。「「血液病学」 (1982年刊) の第3版」
関連項目
- 出血傾向
- 血液凝固障害
- ベルナール・スリエ症候群 - 別の種類のインテグリンの異常によって引き起こされる
- ヴォン・ヴィレブランド病
- 特発性血小板減少性紫斑病